Metagenomic Next Generation Sequencing in clinical Microbiology
Metagenomics is the study of genetic material recovered directly from environmental samples. The broad field may also be referred to as environmental genomics, ecogenomics or community genomics.
Download PDF Brochure of Study, Click Here!
Next generation sequencing is any of several high-throughput sequencing methods whereby billions of nucleic acid fragments can be simultaneously and independently sequenced. Contrast this technique to classical methods such as Sanger sequencing (also known as dideoxynucleotide chain termination sequencing), which processes one nucleotide sequence per reaction.
Metagenomic NGS (mNGS) is simply running all nucleic acids in a sample, which may contain mixed populations of microorganisms, and assigning these to their reference genomes to understand which microbes are present and in what proportions. The ability to sequence and identify nucleic acids from multiple different taxa for metagenomic analysis makes this a powerful new platform that can simultaneously identify genetic material from entirely different kingdoms of organisms.
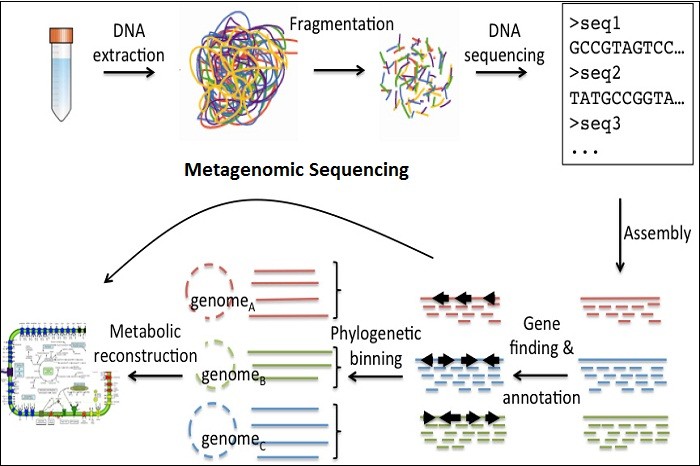
Metagenomics sequencing workflow
There are several steps involved in a sequencing based metagenomics project. These include DNA extraction, library preparation, sequencing, assembly, annotation and statistical analysis.
Sample extraction
A reproducible method to extract DNA from microbial communities is essential for surveying and whole genome metagenomic analysis. Isolation and extraction must yield high quality nucleic acid for subsequent library preparation and sequencing. Sampling variation can have an effect on comparisons, and abundance measurements. This introduces several challenges as some samples must be delivered anaerobically. Exposure to oxygen or freezing can change the dynamic composition of a given microbial community. For example, freezing, thawing and subsequent bead-beating can affect the cell wall of Gram-positive bacteria, and introduce artifacts compared to extraction performed on fresh samples.
Library preparation
One of the biggest considerations for library preparation of environmental samples for shotgun metagenomic sequencing has to do with amplification. Certain types of samples (water, swabs) yield small amounts of DNA, necessitating amplification during library preparation. Amplification by PCR can over amplify certain fragments over others confounding abundance and microbial diversity measurements. Often the user does not have a choice when faced with low inputs of DNA.
Annotation
Once assembled, genes can be predicted and functionally annotated. Genes are typically predicated in one of three ways: 1) de novo gene prediction, 2) protein family classification, 3) fragment recruitment (binning). Functional annotation is performed by classifying predicted metagenomics proteins into protein families using sequence or hidden Markov models (HMM) databases.
Metagenomics, is a field where genomic analysis of microbial DNA from environmental communities is studied. Metagenomic sequencing enables scientists to analyze genes in the given sample. The method enables researchers to assess bacterial diversity and identify the presence of microbes in various environments. Steps involved in metagenomics sequencing include DNA extraction, library preparation, sequencing, assembly, annotation and statistical analysis.
Want to know more about Metagenomic Next Generation Sequencing, click below link..
Get full information and PDF sample of Metagenomic Next Generation Sequencing
Source: theinsightpartners