Узкие места взаимодействия производственной площадки и отдела регистрации лекарственных препаратов: как оптимизировать процессы и снизить затраты на регистрацию?
Я прихожу к директору, я говорю: — Кто сшил костюм? Кто это сделал? Я ничего не буду делать, не буду кричать, я только хочу в глаза ему посмотреть. Выходит сто человек. Я говорю: — Ребята, кто сшил костюм? Они говорят: — Мы! Я говорю: — Кто это «мы»? Они говорят: — У нас узкая специализация. Один пришивает карман, один — проймочку, я лично пришиваю пуговицы. К пуговицам претензии есть? — Нет! Пришиты насмерть, не оторвёшь! Кто сшил костюм? Кто вместо штанов мне рукава пришил? Кто вместо рукавов мне штаны пришпандорил? Кто это сделал? Они говорят: — Скажите спасибо, что мы к гульфику рукав не пришили. Представляете? Их — сто, а я — один. И все стоят, как пуговицы: насмерть. И я сказал: — Привет, ребята! Вы хорошо устроились!
— Аркадий Райкин, миниатюра «Кто сшил костюм?» из сборника «Люди и манекены».
Автор: Ирина Краснова. Заместитель директора департамента развития производства и регистрации OOO «НПФ «Материа Медика Холдинг»
На мой взгляд, миниатюра, выбранная в качестве эпиграфа к статье, очень точно, хотя и несколько утрировано отражает сложившуюся ситуацию при регистрации ЛП.
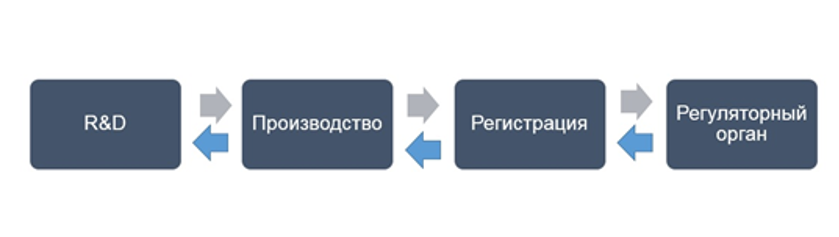
По своей сути, регистрация является «посредником» между R&D, производством и регуляторным органом (рисунок 1). В частности, отдел регистрации осуществляет сбор необходимых документов, формирование регистрационного досье и его подачу в регуляторный орган, сопровождает регистрационное мероприятие и после его завершения предоставляет утвержденные документы производственной площадке.
Давайте разберем основные причины сложившейся ситуации
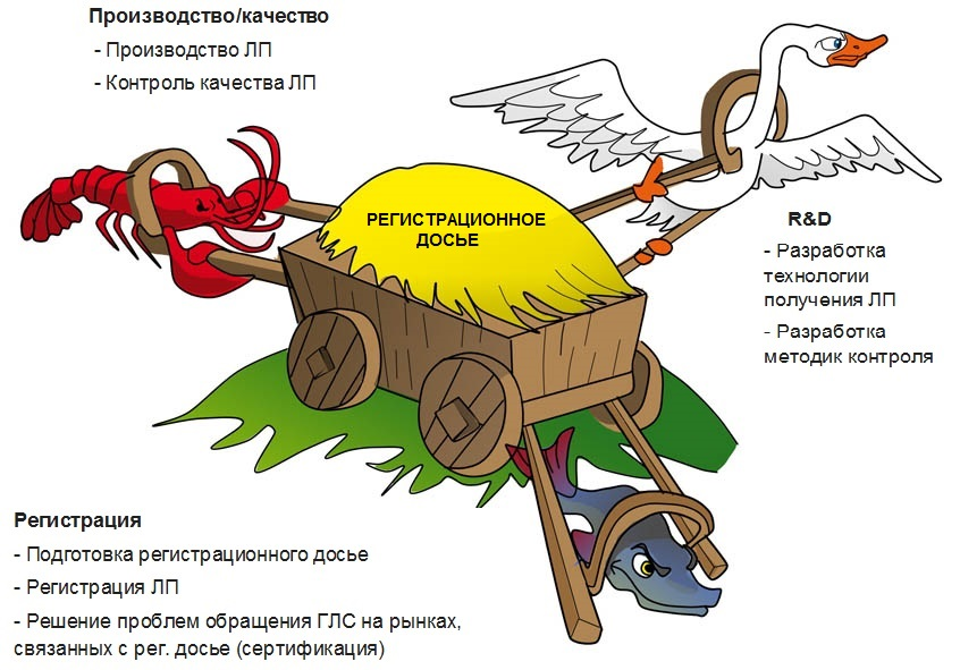
На мой взгляд, одна из причин состоит в том, что у участников есть в некотором роде конфликт интересов, обусловленный противоречивыми задачами, стоящими перед ними (рисунок 2).
Другая причина – административная «оторванность» участников процесса
На рисунке 3 представлена обобщенная организационная структура фармацевтической организации.
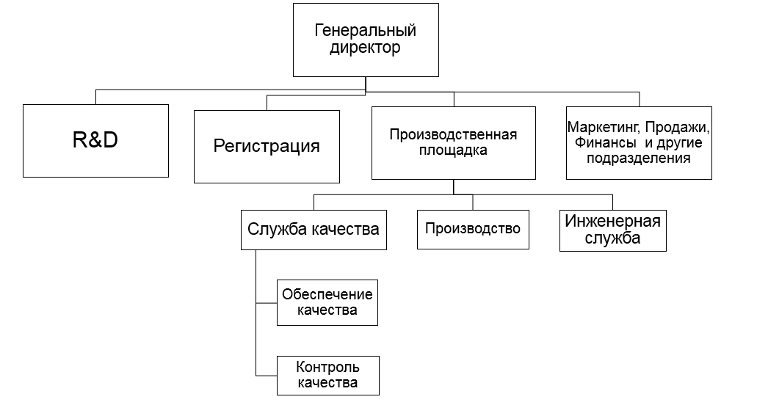
Я намерено не детализировала организационные связи между финансовыми подразделениями, подразделениями маркетинга, продаж и другими подразделениями компании, сделав акцент на участниках процесса регистрации – подразделениями R&D, регистрации и производственной площадке, в составе которой служба качества и производство.
Итак, на что же следует обратить внимание при подготовке регистрационного досье?
Прежде всего, очень важно понимать, что регистрационное досье – это не копия документов производственной площадки.
В связи с этим, при подготовке регистрационное досье необходимо выбрать оптимальную степень детализации. Таким образом, задача заключается в том, чтобы представить всю необходимую информацию в соответствии с регуляторными требованиями, но при этом избежав излишней детализации. Поскольку последняя, с одной стороны, приведет к увеличению трудо-временных и финансовых затрат на поддержание досье в актуальном состоянии, с другой, что менее очевидно, но не менее значимо – предоставляет больший объем информации для экспертизы, что, в свою, очередь увеличивает нагрузку на экспертов и риск ошибок.
Это может быть проиллюстрировано следующей образом: если представить документы производственной площадки и регистрационное досье в виде фотографий (как изображено рисунке 4), то целесообразно выбрать такую, условно назовем ее «степень приближения», которая позволит однозначно идентифицировать объект, но при этом не углубляясь до нюансов, которые не имеют критического значения для качества, эффективности и безопасности ЛП и, как следствие, не значимы при проведении экспертизы документов регистрационного досье.

В этом и заключается виртуозность подготовки регистрационного досье. Для реализации этого необходимо наличие у сотрудников отделов регистрации широкого спектра компетенций, которые не могут ограничиваться только знанием нормативных требований по формированию регистрационного досье и административных регламентов по осуществлению регистрационных мероприятий.
Взаимодействие качества и регистрации осуществляется на следующих этапах:
• Подготовка регистрационного досье
• Выпуск лекарственного препарата в гражданский оборот
• Поддержание регистрационного досье в актуальном состоянии
Ключевые аспекты взаимодействия производственной площадки и регистрации сводятся к следующим:
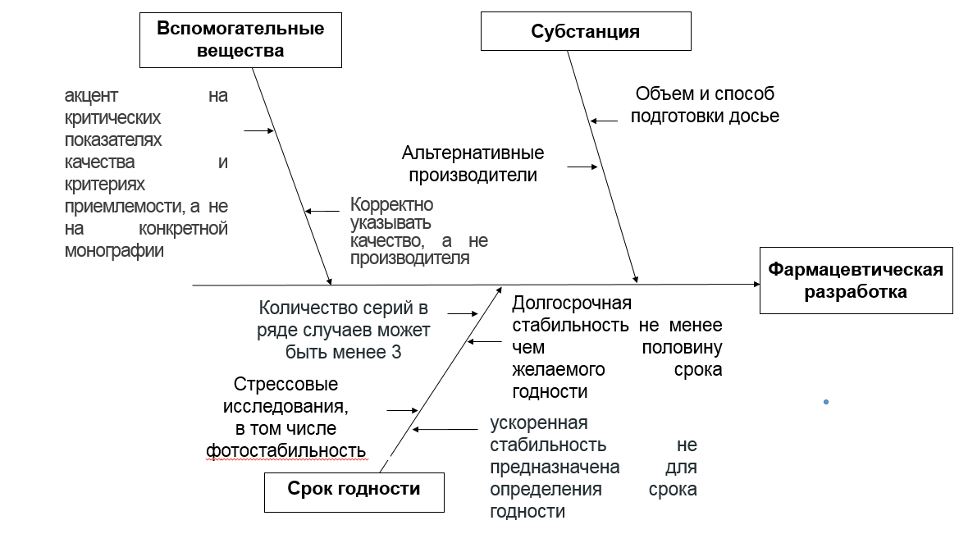
· Состав (АФС и вспомогательные вещества)
· Производство, в том числе контроль процесса
· Срок годности (срок хранения)
Разберем их детальнее, структурировав в соответствии с разделами регистрационного досье.
1.1. Объем и способ подготовки досье в зависимости от статуса и природы АФС, в частности возможны следующие варианты:
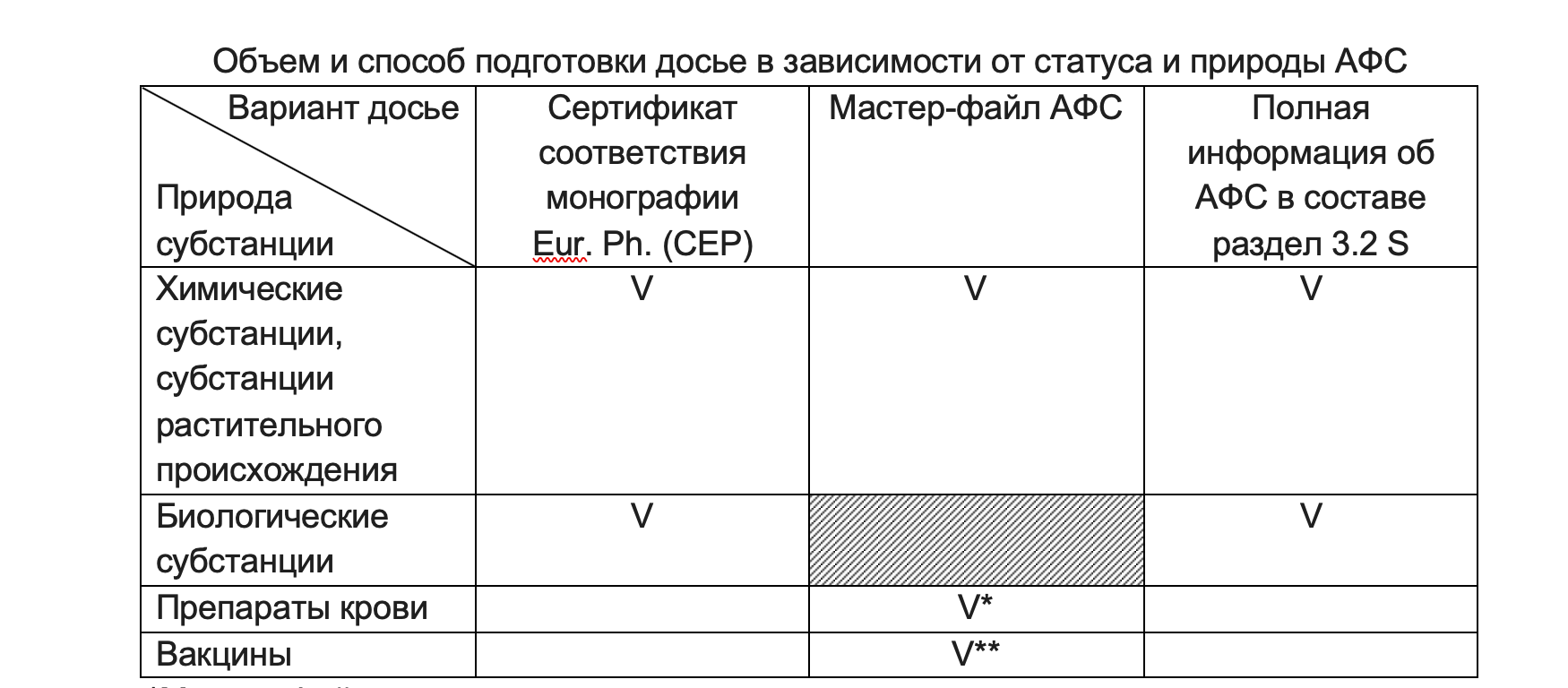
** Мастер-файл на вакцинный антиген
Выбор наиболее оптимального варианта находиться в зоне ответственности заявителя.
Согласно Правилам регистрации и экспертизы лекарственных средств для медицинского применения, утв. Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 78 (в ред. решений Совета Евразийской экономической комиссии от 14.06.2018 № 55, от 30.01.2020 № 9, от 23.12.2020 № 128, от 17.03.21 №36) допускается сокращенный вариант подготовки досье на субстанцию, если она внесена в ГРЛС РФ. К сожалению, не даны четкие инструкции по тому какие документы допускается не предоставлять. В рамках вебинара «Регистрация ЛП по правилам ЕАЭС с учетом новых поправок», организованного Евразийской Академией надлежащих производственных практик 07.04.22 была дана рекомендация предоставлять досье в объеме аналогичном объему при наличии у АФС CEP.
Однако, сокращенные варианты не применимы для ЛП, содержащих в своем составе несколько АФС, а также, если планируется обращение ЛП на территори более одного государства-члена.
1.2. Целесообразно предусмотреть возможность использования АФС от альтернативных поставщиков, что существенно снизит риски возникновения дефектуры препарата на рынке, что может стать особенно критичным в таких фазах жизненного цикла как «рост» и «зрелость».
Корректно указывать качество, а не производителя. При этом при обосновании требований акцент делать не на конкретной монографии, а критических показателях качества и критериях приемлемости. В этом случае не будет проблем при смене производителей вспомогательных веществ, достаточно будет выполнить требования GMP, проведя надлежащим образом квалификации производителей и поставщиков.
3.1. Долгосрочная стабильность должна быть представлена в досье в объеме не менее чем половину желаемого срока годности.
При этом, если в составе досье представлены данные по изучению стабильности. не покрывающие заявленный срок годности, необходимо предоставить в составе досье, соответствующее обязательство по продолжению изучения стабильности.
3.2. Количество серий в ряде случаев может быть менее 3
Более детальную информацию можно найти в Требованиях к исследованию стабильности лекарственных препаратов и фармацевтических субстанций, утв. Решением Коллегии Евразийской экономической комиссии от 10 мая 2018 г. № 69 (в ред. решений Совета Евразийской экономической комиссии от 30.06.2020 N 86).
3.3. Важно, что ускоренная стабильность не предназначена для определения срока годности, необходима для принятия решения в рамках расследования температурных отклонений при хранении и транспортировке
3.4. Стрессовые исследования стабильности, в том числе изучение фотостабильности, исследования стабильности во вскрытом состоянии, в том числе при контакте с пробкой (если применимо) и предоставление результатов в составе регистрационного досье позволит избежать избыточного ужесточения условий хранения. Например, указания «хранить в защищенном от света месте».
Важно продолжать изучение срока годности после регистрации с целью его увеличения, поскольку увеличение срока годности лекарственного препарата принесет значительную экономические выгоды.
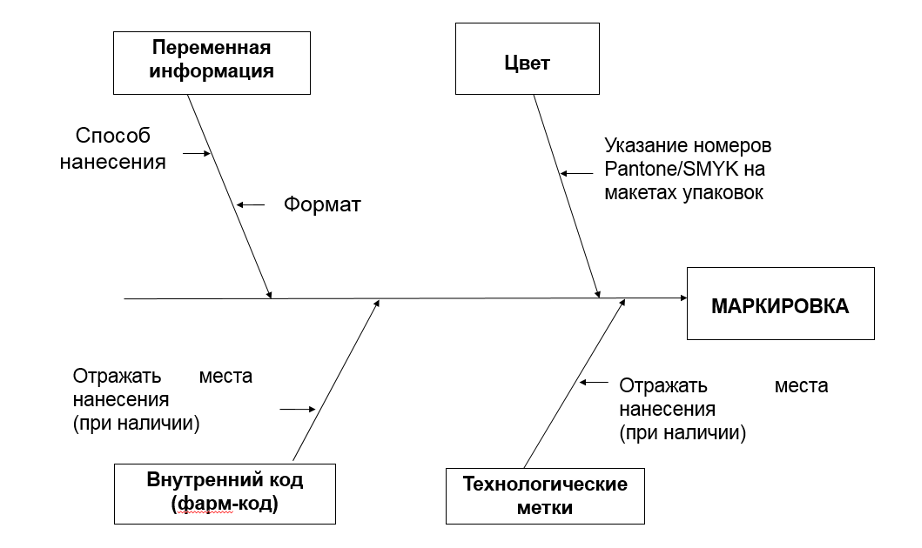
. Целесообразно указание номеров Pantone/SMYK на макетах упаковок при идентификации цветовой гаммы.
Хотя не во всех регионах это жестко регламентировано (примерно в 25% стран, в которых мы работаем), но это существенно снижает риск забраковки серии при независимом контроле, формализует входной контроль упаковочных материалов и претензионную работу с поставщиков (при необходимости), а также упрощает процесс смены поставщика упаковочных материалов.
2. Унифицировать способ нанесения и формат переменной информации
Актуально если площадка выпускает ЛП для различных стран. Это позволит отказаться от перенастройки оборудования, что снизить вероятность возможных простоев, а также снизит вероятность ошибок по причине человеческого фактора.
3. Целесообразно отражение мест нанесения внутреннего кода/фарм-кодов и технологических меток (при наличии), поскольку это существенно снижает риск забраковки серии при независимом контроле.
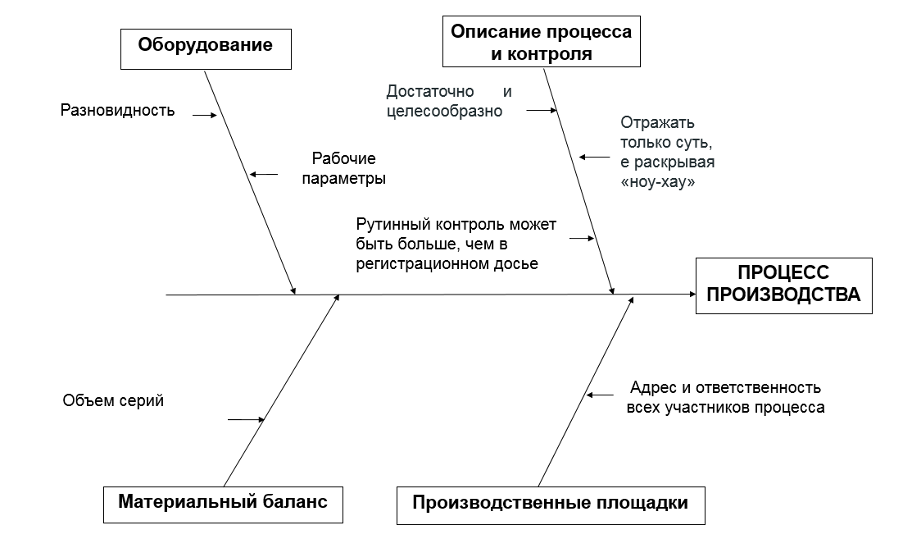
1. Описание процесса и его контроля
Описание процесса и его контроля необходимо проводить в формате достаточно и целесообразно, отражать только суть, не раскрывая «ноу-хау». Важно принять во внимание, что объем контроля в рутинном производственном процессе может превышать таковой, отраженный в регистрационном досье.
Достаточно ограничиться указанием разновидности и рабочих параметров.
3. Производственные площадки и материальный баланс
Необходимо отразить адреса и ответственность всех планируемых участников процесса производства, в том числе выпускающий контроль, а также все объемы серий и фасовок.
Безусловно, это сопряжено с определенными финансовыми и временными затратами на проведение валидационных испытаний и исследование стабильности. Но нужно учитывать, что для определенных объемов серий может проведена валидация процесса производства и изучена стабильность на сериях, часть из которых являются опытно-промышленными сериями. При этом для промышленной серии предоставлен план валидации процесса.
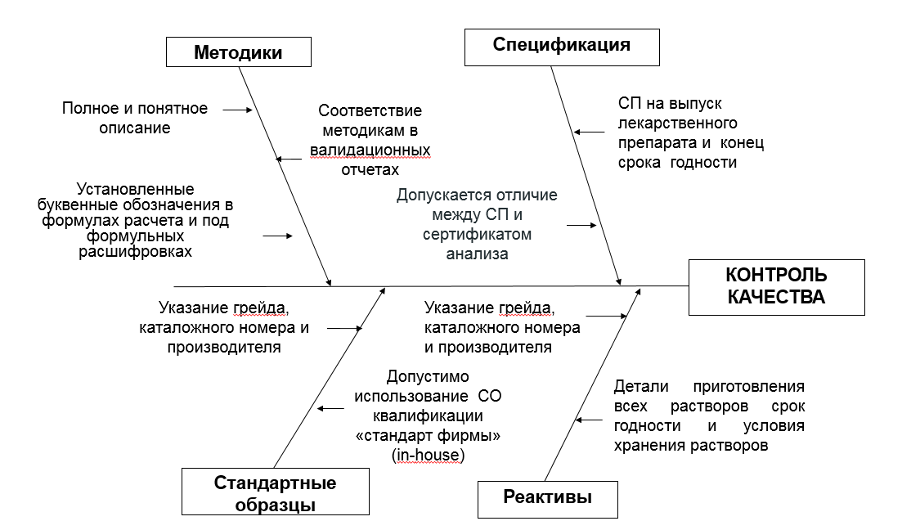
Необходимо комплектовать досье спецификацией при выпуске и спецификацией на конец срока годности (хранения) на основании проведенных испытаний стабильности лекарственного препарата с обоснованием выбора показателей качества, методик испытаний и валидации этих методик, что существенно снизит риски замечаний как при экспертизе документов регистрационного досье, так и в рамках инспектировании на соответствие требованиям GMP.
При том важно понимать, что спецификация при выпуске и спецификация на конец срока годности (хранения) могут отличаться по показателям качества, аналитическим методам/методикам и допустимым пределам. Как правило, допустимые пределы при выпуске ЛП являются более строгими по сравнению с допустимыми пределами на конец срока годности (например, испытание на примеси, испытание на растворение, количественное определение и др.)
Спецификация при выпуске может включать альтернативные методы/методики официально утвержденным (например, ТСХ-ВЭЖХ, СФ-ВЭЖХ).
Показатели качества, используемые во внутрипроизводственном контроле (например, истираемость, твердость, размер частиц и т.п.) включаются опционально.
Различия между спецификацией при выпуске и спецификацией на конец срока годности (хранения) обуславливают допустимость отличий между СП в составе нормативного документа, и сертификатом анализа
Как отмечено выше, нормы, указанные в спецификации (в составе НД), регламентируют качество препарата на конец срока годности, что подтверждено результатами стабильности и, могут отличаться от норм, приведенных в сертификате анализа, указанных на момент выпуска препарата, или отсутствовать.
Описание методики должно быть понятным и содержать всю необходимую информацию для ее воспроизведения (формулы расчета, порядок приготовления калибровочных растворов, значения коэффициентов перерасчета и т.д.)
Крайне важно контролировать соответствие методик, приведенных в проекте НД, методикам, приведенным в валидационных отчетах.
Необходимо использовать только установленные буквенные обозначения в формулах расчета и под формульных расшифровках.
По данным ФГБУ НЦЭСМП Минздрава России от 23.03.22, зафиксирован рост количества невоспроизводимых методик при контроле качества с 13 % до 30%, при этом брак при проведении фармацевтической экспертизы составляет 5%. Эти данные свидетельствуют о плохом качестве подготовки досье, подаче большого количества «сырых» досье.
Реактивы должны быть описаны с указанием грейда, каталожного номера и производителя, лучше указать, если допускается использование аналогичного реактива.
Целесообразно максимально предусмотреть использование фармакопейных реактивов.
Детали приготовления всех растворов (скорость, оборудование, время, температура), срок годности и условия хранения растворов должны быть указаны. Обязательно указание, если используется только свежеприготовленный раствор (если применимо).
Исключение составляют растворы, включенные в фармакопеи, в этом случае допустима ссылка на соответствующую фармакопею. Например, используется 1 % раствор крахмала - в ГФ РФ отражено приготовление только 0,1 % и 0,5 % растворов крахмала, поэтому приготовление 1 % необходимо описать.
Необходимо представить перечень всех используемых стандартных образцов, в том числе стандартов примесей с указанием их квалификации. В отношении квалификации стандартных образцов установлены требования аналогичные требованиям для реактивов. Все стандартные образцы должны быть описаны с указанием грейда, каталожного номера и производителя, лучше указать, если допускается использование аналогичного реактива Целесообразно предусмотреть использование фармакопейных стандартных образцов пригодных для соответствующих методов анализа.
Если нет фармакопейного стандарта, то допустимо использование стандартных образцов квалификации «стандарт фирмы» (in-house), согласно которому данные стандартные образцы используются только для проведения анализов на предприятии и аттестованы по фармакопейному стандартному образцу.
Согласно информации, предоставленной ФГБУ НЦЭСМП Минздрава России от 23.03.22, допускается использование аналогичного оборудования в соответствующих рабочих условиях.
Производить или не производить?
Важным аспектом взаимодействия производственной площадки и регистрации является производство и поставка ЛП после завершения регистрационного мероприятия, частности выбор утвержденных документов для выпуска ЛП. Решающим фактором на данном этапе выступает наличие или отсутствие переходных периодов для производства и поставок.
Возможные варианты развития событий могут быть выбраны при использовании матрицы, представленной на рисунке 9.
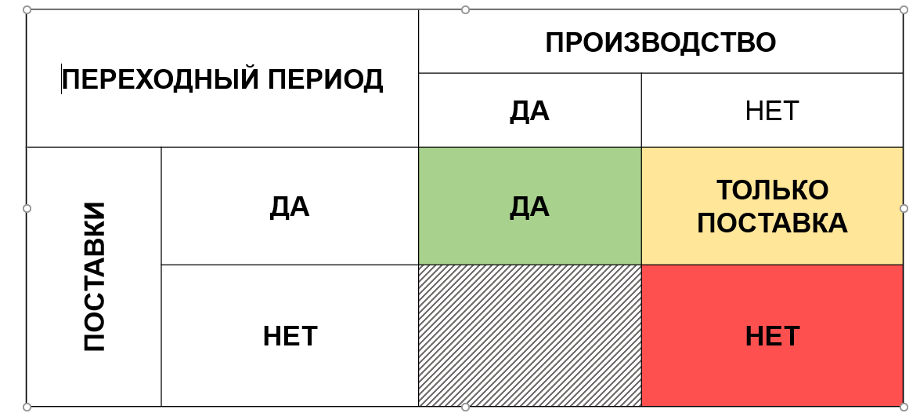
Очевидно, что при наличии переходных периодов для производства и поставок возможны как производство, так и ввоз ЛП по ранее утвержденным документам в течение установленного переходного периода. Прямо противоположная ситуация отмечается в случае отсутствия переходных периодов для производства и поставок.
В случае, если установлен переходный период для поставки, но не предусмотрен для производства, соответственно, возможна только поставка ранее произведенных серий в течение переходного периода.
Однако в законодательстве ряда стран предусмотрены опции, использование которых позволит оптимизировать косвенные затраты на регистрационное мероприятие, в частности обусловленные списанием упаковочных материалов, произведенных в соответствии с ранее утвержденными макетами.
1. В некоторых случаях переходный период для производства действует только при внесении изменений, но не действует при перерегистрации.
Данная ситуация характерна для Азербайджана.
2. Существует возможность производить и ввозить лекарственные препараты по ранее утвержденным документам, даже если срок их действия закончился, но перерегистрация не завершена. Данная опция предусмотрена в Кыргызстане, Молдова.
3. Существует возможность «отложить» дату выдачи новых документов, что дает возможность сработать ранее утвержденные компоненты упаковки и/или поставить уже произведенные по ранее утверждённым документам серии.
Такая возможность законодательно закреплена в Азербайджане и Казахстане.
В течение жизненного цикла любого лекарственного препарата неизбежны изменения, но их количество должно быть оптимальны, и не становиться бумажным и финансовым бременем для компании.
Я не призываю к нарушению нормативных требований, напротив я ратую за их соблюдение, но не в «прямой» трактовке, а путем грамотной профессиональной интерпретации нормативных требований и заводских изменений.
Оптимизация процессов подготовки регистрационного досье и поддержания его актуальном состояниями, и, как следствие, сопряженных с регистрационными мероприятиями финансовых затрат возможна только в результате интеграции регистрации и качества на каждом из вышеуказанных этапов.
Мне представляется целесообразным с одной стороны, использование для формирования регистрационного досье ПО, которое интегрировано с документооборотом производственной площадки.
С другой, необходимо активное участие регистрации в управлении изменениями на производственной площадке с целью формирования адекватной регистрационной стратегии
Особую актуальность этот подход приобретает в настоящее время, когда остро стоит проблема импортозамещения ЛП.
Возможные преимущества и недостатки предложенного подхода представлены на рисунке 10.

👉 Мы в telegram: https://t.me/gxp_center
👉 Наш сайт: https://gxpcenter.ru/
👉 YouTube: https://www.youtube.com/channel/UCrzrxP-5apuDeEoQDJMr1bA